Introduction
Despite a substantial body of published biomarker studies, delirium pathophysiology remains poorly understood and primarily based on animal model data and plausible theoretical possibilities. A biomarker is a biological molecule found in blood, other body fluids, or tissues that is a sign of a normal or abnormal process or a condition or disease.1 They can aid in understanding disease prediction, cause, diagnosis, progression or outcome.2
A recent systematic review evaluating delirium biomarker studies highlighted issues of incomplete and inconsistent reporting of studies.3 The overall low quality of studies limited the reliability and comparability of results and the capacity to synthesise results. This lack of quality reporting norms for delirium biomarker research has likely contributed more widely to heterogeneity of findings and biological and conceptual uncertainty.4
Special configuration for delirium biomarker studies is crucial, as delirium often occurs in the context of other illnesses with overlapping pathophysiological processes; hence, biomarkers of the other illnesses may be inaccurately identified as delirium biomarkers, meaning a binary association is insufficient. The absence of reporting guidelines for delirium biomarker studies has likely contributed to this problem.3
To address this gap, we developed the REDEEMS (Reporting Essentials for DElirium bioMarker Studies), a reporting guideline for delirium biomarker studies.5 This Explanation and Elaboration paper (‘E&E’) is to provide a detailed explanation for each of the REDEEMS guideline items and to promote their implementation.6
Development of the REDEEMS
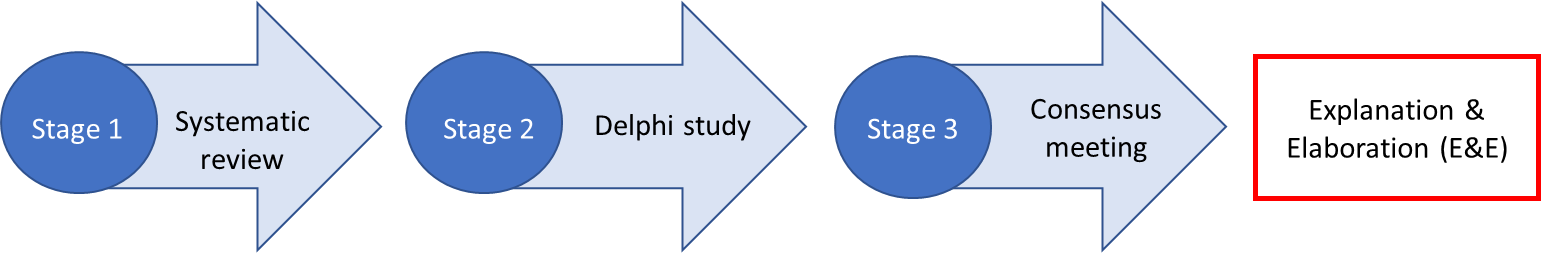
A multi-method, staged process was used to develop the REDEEMS (Figure 1), which is supported by Delphi researchers and those experienced in guideline development6,7 and endorsed by the Equator Network.8

First, a systematic review identified incomplete and inconsistent reporting of many delirium biomarker studies.3 Second, 32 delirium researchers participated in a three-round modified Delphi consensus process of potential reporting items9 informed by the systematic review findings and the REporting recommendations for tumour MARKer prognostic studies (REMARK) checklist.10 Items deemed essential for transparent reporting of delirium biomarker studies by Delphi participants (defined as ≥70% consensus) were included in the preliminary checklist (n=60).9 Lastly, a follow-up consensus meeting was held with a newly configured group of 12 delirium researchers in June 2020 to determine the inclusion or exclusion of 16 items that achieved only borderline consensus (i.e. 70-80%) in the Delphi study.5 Following the meeting, items were further revised and reworded through email correspondence with meeting participants, resulting in the final REDEEMS guideline (Table 1).
How to use REDEEMS
REDEEMS items focus on ensuring transparent and complete reporting of delirium biomarker studies. The REDEEMS guideline does not intend to be a definitive list covering all aspects of delirium biomarker studies. It outlines recommendations for reporting delirium biomarker studies, expecting authors to provide further information and explanation as necessary and according to the specific study design.
The REDEEMS guideline used the REMARK checklist10 as the initial framework before considering additional specifics for delirium biomarkers. REMARK items that the first Delphi round identified as not necessary for adaptation were not re-presented in the second round. In any case these are considered core items for all clinical research reports: i) eligibility criteria, ii) baseline characteristics, iii) recruitment and flow of participants and iv) limitations and future directions. We have not replicated these items in REDEEMS, so we recommend using REDEEMS in conjunction with the most appropriate reporting guideline specific to each delirium study.11
How to use this E&E document
Each REDEEMS item is presented in Table 2, with examples of good reporting from published delirium biomarker literature. A rationale (‘explanation’) for each item is provided (Items 1 to 9). Users should report items according to the individual study or journal requirements, not necessarily in numerical order.
Item 1. Study rationale
A biomarker study aims to explore a biological process and its biological contribution to the clinical event of interest (delirium), possibly as part of a risk/predictive factor analysis or as an effect modifier of outcomes (e.g. mortality). The biomarker under study should be chosen a priori, based on previous data or reasoning that supports a biologically plausible rationale i.e. a testable hypothesis12 and provided early on in the paper.
Given the insufficient biological knowledge of mechanisms underpinning delirium, it is reasonable that the level of justification be hypothetical until more data on its pathophysiology emerges. It is important to note that not all delirium biomarker studies will study a hypothesis, and it is also reasonable to conduct a delirium biomarker study using an exploratory approach. If the study is not testing a specific hypothesis, it should be made clear the study is undertaking an exploratory or ‘un-biased’ approach.
For some research questions, a control or comparator group will be needed to test the hypothesis, and the choice should be clearly justified. Control or comparator groups to consider in a delirium biomarker study include participants without delirium, healthy participants, and/or participants with the same underlying diagnosis and/or illness severity without delirium. In longitudinal studies, the group under comparison may include participants without delirium, or delirium with shorter delirium duration and/or lower severity.
Item 2. Ascertainment of delirium
There is vast variation in how delirium is assessed, including subjective clinical judgment, structured instruments, and comprehensive processes supported by cognitive testing.13 Standardisation of process and reference rater characteristics will help to ensure more reliable assessment of delirium cases and severity,14 and comparability of results. Delirium must be ascertained using a structured tool or process for which psychometric properties have been established (e.g. reliability, validity, discriminatory power, and normative data).4 This is only likely to be achievable in the context of a prospective study.
Item 3. Outcome measures
Relevant endpoints need precise definitions. For example, it is insufficient to refer to the endpoint as ‘delirium severity’ without reporting how severity was measured. Wherever possible, standardised definitions are also recommended. Blinding is particularly important if the endpoint is potentially subject to measurement bias (e.g. delirium severity), while less important for definitive endpoints such as death.15 Reporting whether and how the analyser was blinded to outcomes, allows the reader to assess the risk of measurement bias.
Item 4. Assay procedures
These items were derived from the REMARK checklist10 but included in the REDEEEMS guideline as they have been identified as a priority area for improvement in reporting.3
Detailed reporting of assay methods allows others to assess their adequacy, replicate it with precision and accuracy, and report any potential limitations that may impact interpretation of results. If another widely accessible document detailing the exact assay method is used (for example, a commercially available assay protocol), it is acceptable to cite that document. If a commercially available kit is used for the assay, it is important to state whether the instructions were followed and, if not, explain any deviations from recommended procedures.
Despite complete standardisation of the assay and quality monitoring, random variation (measurement error) in assay results can still occur due to assay imprecision or variations across laboratories. Therefore, reporting strategies such as taking the average of two or three results to produce a measurement with less error, are preferred. Reporting reproducibility assessments provide a sense of the overall variability in the assay. Batch effects should also be considered.16
It is vital to include as much detail as possible about the type of biological sample used in the study and how it was collected, processed, and stored. The time of specimen collection often will not coincide with the assay, e.g. after a period of storage. Storage timings, durations and conditions, such as temperature, should be reported as standard. Serum or plasma specimens should report collection methods, including anticoagulants used, temperature before storage, the storage tube type, processing protocols, and preservatives used. The Biospecimen Reporting for Improved Study Quality (BRISQ) guideline provides detailed recommendations on what should be reported for specimen collection, processing and storage when publishing research biospecimens.17
Objective measures are not subject to a large degree of individual interpretation and are likely to be reliable across patients.18 However, a patient’s clinical outcome is sometimes known by the individual running the assay and analysing the results, which can increase the risk of measurement bias. Reporting the extent of blinding of the assay assessor to clinical outcomes allows assessment of the risk of this type of bias.
Item 5. Timing of collection of the biological sample
Different phases of delirium are associated with varying biomarker findings.19 Therefore, a thorough description of the timing of specimen collection in relation to onset, presence, and resolution of delirium is critical. In populations with prevalent delirium, or at risk of incident delirium, longitudinal samples are an advantage.
It is important to justify sample collection timing according to the clinical scenario and/or tested hypothesis. For example, clinical insults (surgery, anaesthetic); clinically relevant decision points (e.g. extubation, discharge); when the delirium precipitant is likely to have resolved; or based on biomarker kinetics, such as after sepsis when an inflammatory biomarker is likely to change. This reporting allows the reader to make an informed judgement of the appropriateness of the timing of biomarker collection, while more consistent overall reporting will promote better understanding of associations between clinical, delirium and biomarker trajectories.
Item 6. Confounding variables
Multiple aetiologies cause delirium in heterogenous groups of clinical populations with varying baseline risk factors, all of which could confound the delirium-biomarker relationship.20 Imprecise or unmeasured variables can increase the risk of residual confounding.21,22 The confounding variables should be based on known relationships with the outcomes of interest or help define subgroups of interest within the population. Dementia status (and how it was ascertained) is essential to collect, as it is the strongest risk factor for delirium and biomarkers of delirium and dementia overlap. Other important confounders include age, baseline cognitive impairment and severity of illness.
Item 7. Sample size
Underpowered studies limit the ability to detect true differences in biomarker findings and to draw firm conclusions. For example, if a study with negative findings is inadequately powered, a clinically important but statistically non-significant effect could be erroneously ignored.23 Sample size should be determined based on the estimated effect size of the biomarker in predicting the outcome. The estimated incidence or prevalence of delirium also needs to be considered. Sample size calculations should ideally be decided a priori based on previous studies or pilot data where possible.
Item 8. Statistical analysis
Many studies will have missing biomarker or covariate data. Authors should report missing data for each clinical variable, detail its nature (missing at random (MAR), missing completing at random (MCAR) and report how the missing data were handled (case-wise deletion, multiple imputation, etc.). The statistical plan should account for biomarker missing data due to clinical attrition, such as overall deterioration, worsening cognition and death, all of which are common in delirium studies. Missing data due to the practical challenges of biomarker collection in people with delirium should also be reported. These include situations where a patient refuses specimen collection, is at a procedure or is too sick for collection. Missingness may differ depending on whether the study is cross-sectional or longitudinal. Biomarker concentrations under the limit of detection might be assigned a value equivalent to 50% of the detection limit, or else a concentration of zero where non-detection can reasonably be assumed to reflect a true absence.
Item 9. Univariable and multivariable analysis
Results should be reported for all primary and secondary endpoints, ideally following a pre-specified, registered analysis plan.24 The unadjusted and adjusted results should be reported together. For adjusted analyses, the number of included participants should be reported if this differs because of missing covariate values.
For each outcome, the study should report a summary outcome between groups as an estimated effect size). For binary outcomes, the estimated effect size could either be the risk ratio (relative risk), odds ratio, or risk difference. Confidence intervals (CI) should also be presented for all outcomes to indicate the precision of the estimate.25,26 CIs are particularly important for differences that did not meet a statistical significance without ruling out a meaningful clinical difference.27 P values should not be reported in the absence of CIs.
It is helpful to report on univariable and multivariable results, allowing for a direct assessment of how the biomarker is altered by including standard covariates in stepwise adjustments. Authors should report all potential confounding variables and the criteria for including or excluding variables in multivariate models. Decisions about excluding or including variables should be guided by knowledge, or explicit assumptions, on causal relations. Careful consideration of biomarkers that are confounders versus those that are mediators is important. Inappropriate decisions may introduce bias; for example, by including confounding variables in the causal pathway (i.e. mediators) that occur due to an underlying illness such as sepsis. Inappropriate adjustment for sepsis in this example may lead to an adjustment for variables in the causal pathway.12
Conclusion
Poor quality of reporting of delirium biomarker studies is a barrier to developing knowledge of delirium pathophysiology. The REDEEMS guideline seeks to address this barrier by providing items specific to reporting delirium biomarker studies.
The REDEEMS guideline was developed to guide authors in reporting delirium biomarker studies transparently. Good reporting of studies will increase the potential for synthesis of studies through meta-analysis. The guideline will help researchers to be more informed of the critical elements of a delirium biomarker study and they can be applied from the initial process of study design, through to the conduct, analysis, and ultimate reporting. While we acknowledge that it may not be possible to fulfil all reporting requirements from the guideline, we encourage authors to assess the impact of missing information, and report the rationale for its absence, when possible.
The REDEEMS guideline and E&E paper were developed as a collaborative effort of delirium researchers committed to improving the understanding of delirium pathophysiology. The next step is the dissemination of the REDEEMS and E&E to promote uptake.6 Authors of future delirium biomarker studies can contribute to transparent and complete reporting by using the REDEEMS guideline and recommending it to others in the field. As new evidence emerges and critical feedback is obtained, the REDEEMS will be updated.